What We Do Here
Changes Lives Everywhere
Explore why Dana-Farber is a leading-edge cancer center, and how our discoveries and innovative approaches to care are transforming cancer treatment all over the world.

Discover the Dana-Farber Difference
Our Momentum of Discovery
Dana-Farber has led the way in cancer breakthroughs for more than 75 years. Our national advertising campaign highlights our momentum of discovery and shows how what we do here changes lives everywhere.

Groundbreaking Research
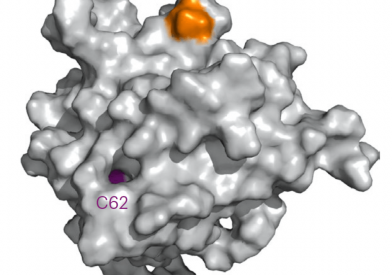
Dana-Farber Cancer Institute remains true to Dr. Sidney Farber's vision of a cancer center that is as dedicated to discoveries in cancer research as it is to delivering compassionate, patient-centered care. Discoveries made by Dana-Farber researchers today become tomorrow’s breakthrough treatments.

New England's #1 Cancer Center
Dana-Farber Brigham Cancer Center has been New England's top-ranked cancer center for over 20 years. Because we specialize in cancer, we're alert to every aspect of treatment and care that can help you get well.
Everything we do is centered around you.
How You Can Help

Support Dana-Farber Cancer Institute and The Jimmy Fund's mission to prevent, treat, and defy cancer.




News and Discovery





















Innovation and Insights