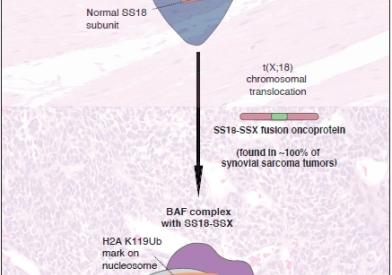
What Is Synovial Sarcoma?
Synovial sarcoma, a malignant soft tissue tumor often located in the extremities, is so named because it exhibits a microscopic resemblance to synovial tissue, the lining found within joints, though it is not thought to originate from this tissue.
Predominantly affecting adolescents and young adults, synovial sarcoma displays a higher occurrence in males. While its primary site is in the extremity, it can manifest in many locations throughout the body. The tumor has the potential to metastasize, particularly involving regional lymph nodes. Distant tissue spread occurs in approximately half of all cases, often months to years post initial diagnosis, though it can also be present at the time of diagnosis.
Synovial sarcoma is classified as a rare tumor, categorized among soft tissue sarcomas — cancers originating in soft tissues such as fat, muscles, tendons, nerves, blood vessels, synovial tissue, and fibrous tissue. Collectively, soft tissue sarcomas represent less than 1 percent of annual new cancer cases. In the United States, this accounts for roughly 900 instances among children and adolescents.
How We Treat Childhood Synovial Sarcoma
Children diagnosed with synovial sarcoma receive care at Dana-Farber/Boston Children's Cancer and Blood Disorders Center through our dedicated Bone and Soft Tissue Tumors Program. This comprehensive pediatric oncology service seamlessly combines the exceptional knowledge of a leading cancer center with a renowned, world-class children's hospital. Within this specialized program, we provide a complete array of treatment options for bone and soft tissue tumors, with our accomplished solid tumor treatment team working with you to decide the best approach for your child's needs.
Our esteemed pediatric oncologists not only have access to but also take the lead in pioneering cutting-edge clinical trials tailored for synovial sarcoma. Our surgeons are very experienced in the management of synovial sarcoma, and moreover, our radiation oncologists are equipped with the latest advancements in radiation therapy techniques, enhancing the potential care avenues for children affected by synovial sarcoma.