New technique to determine protein structures may solve biomedical puzzles
‘Experimental evolution’ analyses can determine the interactions that proteins use to fold into functional three-dimensional shapes.
“Proteins are the workers in the cell, and it's important to know their shape,” says Chris Sander, PhD, director of Dana-Farber’s cBio Center in the Department of Data Sciences. Sander and his colleagues have now demonstrated a powerful “experimental evolution” method to discover details of protein shape and function, and the method may find uses across a very broad spectrum of biomedical research.
“This is a basic discovery in molecular evolutionary biology, with possible applications to cancer,” says Sander, professor of cell biology at Harvard Medical School and co-senior-author on a paper describing the work in the journal Cell Systems.
Among its potential applications, the “3Dseq” technique may aid in determining the three-dimensional structure of cancer-related proteins that haven’t been identified by alternative methods. The new technique also may help in understanding how oncogenes and tumor suppressor genes evolve in cancer, and in pinpointing which mutations in those genes contribute to disease progression.
For decades, molecular biologists have known that a protein’s ability to function in a cell relies on its proper shape, which is dictated by the order of its constituent amino acids. Determining protein three-dimensional structure, however, requires complicated experimental techniques.
Back in 2011, working with Harvard associate professor Debora Marks and other colleagues, Sander took a big step forward on the challenge of predicting protein structure using a mathematical approach based on evolution. The “natural evolution” approach begins by analyzing how the genetic sequence of a particular protein changes over millions of years. To do this, the team examined the sequence of the protein across evolutionary time—from ancient species such as bacteria to more recently evolved species such as mice and humans.
The key insight in the earlier study came when the team used computational methods to identify which amino acids in the protein are interacting with each other, by looking at amino acids that change in lockstep across evolution.
“We look at things that co-vary, where if one thing changes in the sequence, another thing changes,” Sander says. “It’s like a nut and a bolt—if you change one part, you have to change the other part so it still fits.”
He and his colleagues came up with a basic mathematical trick that can find changes in the amino acids that produce a direct effect on protein structure, providing key information that can be fed into existing algorithms from molecular physics to compute the structure.
However, not all proteins can be studied using sequences found in natural evolution. The latest innovation from the Sander group is to bring evolution into lab dishes, where the process can be tightly controlled and take weeks rather than millions of years.
Dana-Farber research scientists Michael Stiffler and Frank Poelwijk are co-lead authors, and Nicholas Gauthier is co-senior author, on the new paper about the project, which is the first-large scale demonstration of an experimental evolution method to determine protein structure.
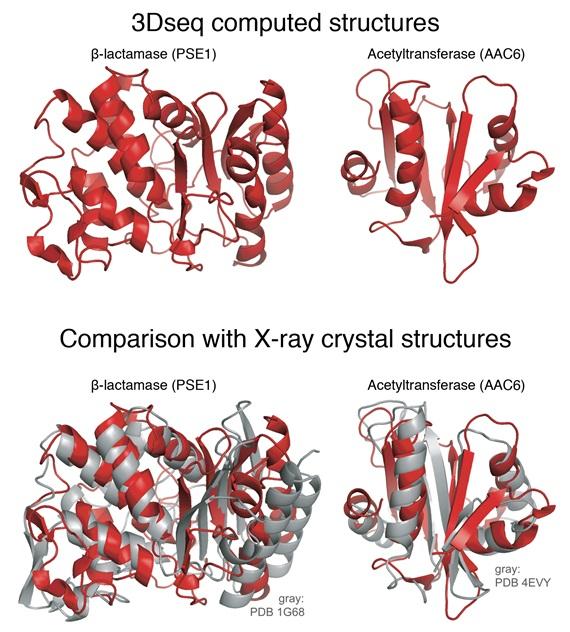
The scientists started with a gene for an enzyme from E. coli bacteria that renders bacteria resistant to a common antibiotic. First they generated millions of copies of the original gene with mutations sprinkled in various positions, and then put these mutated genes into millions of bacteria. They then put an antibiotic into the lab dishes holding the bacteria, and harvested the bacteria that survive. These surviving bacteria have functional antibiotic resistance genes yet still contained selected mutations. This painstaking procedure was repeated many times to mimic evolutionary processes in analogy to natural evolution. “Out of tens of millions of proteins, we ended up with a few hundred thousand that actually work,” Sander says.
Using the computational strategy pioneered in the 2011 study, they produced data that generated 3D protein structures for two very different antibiotic resistance proteins and got shapes very similar to the ones determined by X-ray crystallography.
3Dseq experimental evolution will join three existing technologies to reveal protein structure: X-ray crystallography, which shoots X -rays at a crystallized protein; nuclear magnetic resonance (NMR) spectroscopy, based on physics similar to that employed by magnetic resonance imaging (MRI) scanning; and cryo-electron microscopy, which scans frozen specimens with an electron microscope.
Once it is mature, the 3Dseq technology may bring two main advantages to protein structure determination, Sander says. First, the three existing methods do not always work for proteins. Second, 3Dseq provides details on the key interactions in the complicated protein shapes that are required for proteins to function. This capability eventually could prove to be very important for a number of applications in cell biology, ranging from understanding how pathogens evolve to accelerating the development of biotherapeutics, he says.
His group and its collaborators have begun efforts to improve the 3Dseq screening techniques and generalize the technologies for use with other proteins. Their paper was published along with its sequence data and software tools. “We will collaborate with other people to develop the assays to make it more generally applicable to proteins of interest,” Sander says. “Whatever we develop, we’ll make available publicly.”
“This research shows the positive, open-minded scientific culture of Dana-Farber and Harvard Medical School, as an example of basic science that will spawn advances in cancer research,” he adds. Lead funding for the work came from Dana-Farber and the National Institute of General Medical Sciences.
Media Contacts
If you are a journalist and have a question about this story, please call 617-632-4090 and ask to speak to a member of the media team, or email media@dfci.harvard.edu.
The Media Team cannot respond to patient inquiries. For more information, please see Contact Us.