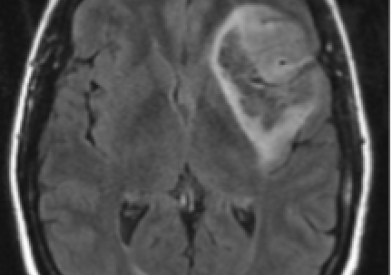
What Are Brain Tumors?
A tumor is a mass of tissue made up of cells that grow and multiply in an abnormal and uncontrolled way. The brain controls many important bodily functions; when a tumor grows into or presses on an area of the brain, it may stop that part of the brain from functioning normally.
A tumor is a mass of tissue made up of cells that grow and multiply in an abnormal and uncontrolled way. The brain controls many important bodily functions; when a tumor grows into or presses on an area of the brain, it may stop that part of the brain from functioning normally.
A tumor may be either benign (not cancerous) or malignant (cancerous); both cause symptoms that require treatment:
- Benign brain and spinal cord tumors grow more slowly, but can press on nearby areas of the brain. They rarely spread into other tissues, and may recur (come back) after treatment.
- Malignant brain and spinal cord tumors are likely to grow quickly and spread into other brain tissue.
Brain tumors are classified as primary or secondary:
- Primary brain tumors develop inside the brain or spine, and almost never spread outside of the brain or spine. Primary brain tumors are most commonly named after the cells from which they derive. Some types of primary brain tumors include:
- Glioblastoma
- Glioma
- CNS (central nervous system) Lymphoma
- Meningioma
- Secondary brain tumors are tumors found in the brain that have started somewhere else in the body. These are also known as metastatic brain tumors, or brain metastases. Metastatic brain tumors are more common than primary brain tumors.