Dana-Farber study advances understanding of rare sarcoma
- Scientists discover how abnormal protein disrupts gene expression in synovial sarcoma
- For the first time, scientists discover the molecular basis for the cancer-specific targeting properties of the culprit fusion protein found in synovial sarcoma
- Findings could help with drug development to treat the rare cancer
Advancing the understanding of synovial sarcoma, a highly aggressive and rare cancer of young people, scientists at Dana-Farber Cancer Institute have discovered how an abnormal protein misdirects and disrupts the control of gene expression in cells, causing the aberrant activation of normally repressed genes that contribute to the malignant growth of sarcoma tumors.
The investigators, led by Cigall Kadoch, PhD, say the new insight could guide efforts to develop drugs to treat synovial sarcoma, which primarily affects muscle tissues in the extremities. The root cause of the sarcoma is a cellular accident — the abnormal joining of two proteins, resulting in a “fusion protein” that sets in motion the malignant malfunctions.
“We have for the first time defined the molecular basis for the cancer-specific targeting properties of the culprit fusion protein, SS18-SSX, found in 100% of cases of synovial sarcoma.” Kadoch said. “Importantly, this provides a powerful new therapeutic identification strategy for the drug discovery industry” to develop drugs to block the interaction between the abnormal fusion protein and nucleosomes — structures that package and organize DNA in cells, said Kadoch, senior author of the study published today in Nature Structural and Molecular Biology.
Nucleosomes, spool-like structures resembling beads on a string, make up chromatin, a term to describe DNA wrapped around proteins called histones. Chromatin is found throughout the genome, which in humans contains an estimated 20,000 genes and much other genetic information. Modifications in chromatin structure, carried out by protein complexes called chromatin remodelers, regulate the turning on and off of genes for cell- and tissue-specific purposes. Malfunctions of the chromatin remodeling system have been implicated in many cancers and neurodevelopmental disorders, a major focus of the Kadoch laboratory.
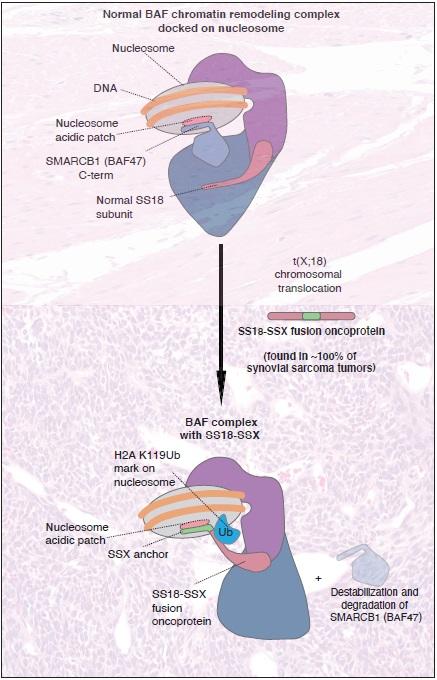
In synovial sarcoma, the SS18-SSX fusion oncoprotein replaces the normal SS18 subunit, or component, in BAF chromatin remodeling complexes. As a result, BAF chromatin remodeling complexes are “dragged around the genome to all the wrong sites” on chromatin “to aberrantly activate them and drive the gene expression and proliferation of synovial sarcoma,” Kadoch and her team previously showed in a 2018 study. These “wrong” sites, she explained, are regions on chromatin marked by polycomb repressive complexes, which normally keep genes such as stem cell genes from being expressed when not needed. One might think of a complex traffic system in which millions of aircraft or ships normally proceed to specified destinations along specific routes – but in the case of some cancers, the targeting of these vessels is scrambled, with disastrous results.
However, this previous work had not revealed how the SS18-SSX fusion protein and BAF complexes were erroneously targeted to those repressed sites across the genome: “it was a mystery until now,” Kadoch said.
In the experiments described in the new paper, Kadoch and her co-authors found that a minimal, small segment of the SS18-SSX fusion protein binds to the so-called “acidic patch” on nucleosomes. The acidic patch is a pocket of negatively charged molecules to which chromatin remodeling complexes, such as BAF complexes, can dock. The researchers found that the small segment of SSX – which they refer to as the “tail” of the fusion protein – is made up of only 34 amino acids, but binds extremely tightly to the acidic patch. Specifically, the SSX tail binds to the acidic patch on nucleosomes that are “decorated” with a histone modification known as ubiquitination of histone H2A (H2AK119Ub).
In fact, the tail of SSX binds so powerfully that it dislodges a BAF complex subunit known as SMARCB1 (BAF47) that normally engages the complexes on chromatin. “These complexes are really stuck, so once they get to the site, they stay there and strongly activate those cancer-related genes,” noted Kadoch.
According to the researchers, the full-length SSX protein is normally only active in the testes, where it likely plays a role in sperm development. But when a part of the SSX protein is fused with the BAF complex SS18 subunit (via the characteristic t(X;18) chromosomal translocation in synovial sarcoma), it prompts an “unfortunate scenario” that leads to the remodeling or opening of chromatin over the regions where the SSX tail guides BAF complexes via the acidic patch, causing the activation of cancer-related genes
With these new insights into how the oncogenic fusion protein targets BAF chromatin remodeling complexes on the genome leading to activation of synovial sarcoma-related genes, said Kadoch, “these results suggest that inhibition of the interactions between SS18-SSX-bound BAF complexes and H2AK119Ub -marked nucleosomes using small molecules or peptides may prove a viable therapeutic strategy for synovial sarcoma.”
Co-first authors on the study are Matthew McBride, Ph.D., a recent graduate of the Harvard Chemical Biology program, and instructor and research fellow, Nazar Mashtalir, Ph.D.
This work was supported in part by awards from the NIH DP2 New Innovator Award 1DP2CA195762- 01 (C.K.), the American Cancer Society Research Scholar Award RSG-14-051-01-DMC (C.K.), the Pew-Stewart Scholars in Cancer Research Grant (C.K.), Alex’s Lemonade Stand Foundation ‘A Award’ (C.K.), NIH 1R01 CA237241-01 (C.K.), the NIH Cancer Moonshot FusOnc2 1U54 CA231638-01 (C.K.), NIH K99/R00 K99CA237855NIH (N.M.), and R37-GM086868 (T.W.M.) and NIH PO1-CA196539 (T.W.M.).
Kadoch is the scientific founder, fiduciary board of directors’ member, scientific advisory board member, shareholder, and consultant for Foghorn Therapeutics, Inc (Cambridge, MA).
Media Contacts
If you are a journalist and have a question about this story, please call 617-632-4090 and ask to speak to a member of the media team, or email media@dfci.harvard.edu.
The Media Team cannot respond to patient inquiries. For more information, please see Contact Us.